Structure and function of bulk proteins of myocardial microfilaments
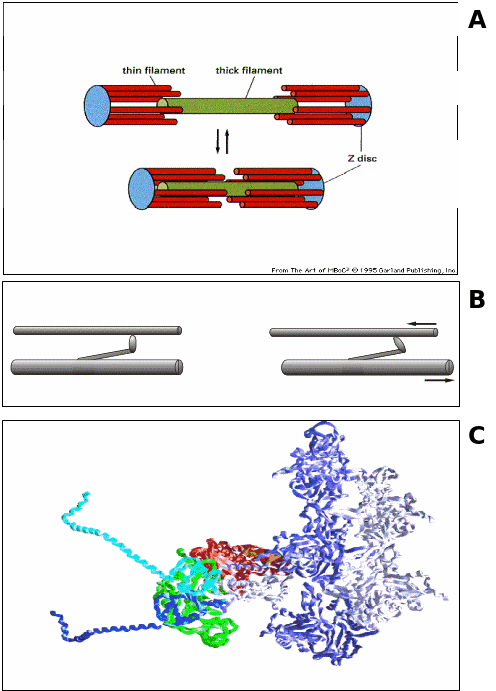
The cellular units of cardiac muscle fibers, myocytes (10-20 µm diameters and 50-100 µm length) are connected end-to-end in series by a network of interdigitating cell membrane called intercalated discs, comprising two units, the desmosomes and gap junctions. These intervening structures not only hold the fibers together, they also allow the action potential to run over the entire length of connecting fibers. Myocytes surrounded by cell membrane, sarcolemma, possess, besides the usual cell inclusions, compact bundles of longitudinally arranged myofibrils. Each myofibril is composed of small repeated units called sarcomeres that are joined from the ends and are poised between two adjacent Z-lines. The myofilaments comprise ca. 45-60% of net cardiomyocyte’s volume. The sarcomere gives striated appearance, and is composed of the muscle contractile proteins categorized into thin or principally actin, along with several regulatory and organizational proteins, and thick or myosin mycofilaments. Figure 1.1 shows the anatomical structure of sarcomere.
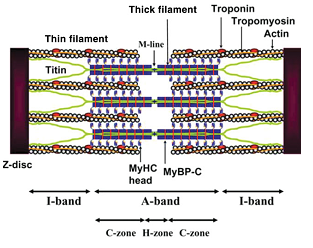
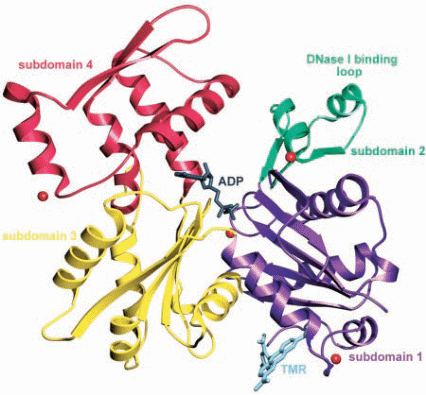
As major share of thin filament, two inter-winding helical chains of monomeric G (globular)-actin proteins prevailing at low salt concentrations, upon polymerization at physiological salt concentrations form a filamentous macromolecule of F (fibrous)-actin with thickness of 10 nm and average length of 1 µm. F-actin is a twisted rope made up of G-actin monomer strands with a half pitch cross over every 35.5 nm (Figure 1.2). Thin filament constituents are also two classes of proteins involved in contraction and signal transduction, called tropomyosin (cTm) and troponin (cTn). cTm which is ~40 nm in length stays aligned to the backbone of the actin protein. The thin and the thick filaments slide over each other in a typical arrangement and together they constitute a zone termed as A-band. Whilst the thin filament lies in between the myosin thick filaments, it protrudes towards the margins and remains partially anchored in the Z-line that forms the I-band. About 22% of muscle protein is constituted by actin. Much of the structural information of actin myofilaments was obtained from X-ray crystallographic analysis, which shows presence of four sub-domains per actin molecule (Figure 1.2). These sub-domains in the native states are separated by a cleft that contains either Mg2+ or Ca2+ and the bound ADP, thus providing a linker for stable association of the sub-domains (Kabsch, Mannherz et al., 1990). The crystal structure further indicates that the exterior sub-domains 1 and 2 that interact with myosin constitute the surrounding structure of the filaments. The positions of sub-domains 3 and 4 are more centralized, projecting towards the helical axis, where they associate with identical sub-domains of adjacent actin monomers. The sub-domain 1 contains both the N-terminal and C-terminal ends of actin monomers (Holmes, Popp et al., 1990). This is probably the most conserved protein among all the muscle proteins (only 5% divergence in different species; Pollard, 1990). Six isoforms of actin are reported to be expressed in mammalian cells, grouped into three isoforms, α-, β- and γ, each for the cardiac and the skeletal cells. In myocardium predominantly the α- isoforms of both cardiac and skeletal actins co-express (Morimoto 2008).
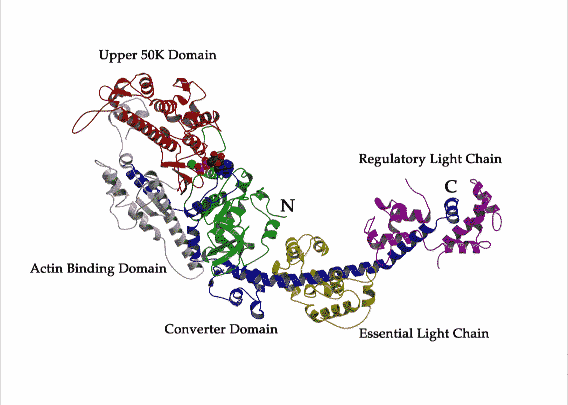
The thick filament is another coiled macromolecule of approximately 300 myosin residues (mass, ~500 kDa), besides the non-contractile proteins like titin and myomesin that contribute to the stiffness of muscle fibers. Each myosin myofilament is approximately ~1.6 µm long and ~15 nm wide. Electron microscopic analysis suggests that myosin has a double headed globular protein attached to a long rod. The rod is a two-stranded α-helical coiled coil. Cardiac isoform of myosin binding protein C is a thick filament protein that, within the sarcomeric A-band region, spans over myosin myofilaments at regular intervals of 43 nm. The presence of this protein subdivides the A-band into two additional segments, C- and H-zones. When myosin molecule is cleaved with trypsin, two fragments are attained, thinner and slender light meromyosin (LMM) of ca. 100 nm length, and globular and shorter heavy meromyosin (HMM). The HMM can be again proteolytically cleaved with papain into sub-fragment S1 (20 nm) and sub-fragment S2 (50 nm). Each of the two S1 sub-fragments is comprised of one heavy chain (400 kDa) and a pair of light chains (each 20 kDa). S1 globular subunit functions as “head” where the ATP hydrolysis and cross-bridging take place, while the S2 serves as “arms” which extends to LMM. The S1 head is composed of the ATP and actin binding sites and an extended α-helical C-terminal “tail” constituted by S2 and portion of LMM (Rayment, 1993 #562}{Xie, 1994 #536). Morimoto (2008) reported that there exist two isoforms of cardiac HMM, α and β, constituting the S1 “heads”. It has been shown that in small mammals like rodents, α- isoform is the predominant atrial and ventricular HMMs throughout the adulthood, whereas β-isoform is expressed in the ventricles and α-isoform in atria in case of adult larger mammals like rabbit and human. Incidentally, the β-HMM is also found to be expressed in slow skeletal muscle type I fibers. In our studies we have used S1 “head” and Figure 1.3 is a representation of the 3-D crystal structure of a skeletal muscle S1 “head”.
Tropomyosin
Tropomyosin is a component of thin mycofilament which adheres to actin. It is a right-handed helical protein which forms a coiled dimmer. Although isoform diversity with respect to the tissue of origin and functions exists, most of the tropomyosin exhibit remarkable conserved phylogeny (Gunning et al., 2005). In human at least two genes code for two variants of tropomyosin, α and β. In fast skeletal muscles mixed α-, β-, and in cardiac muscles only α-, α- isoforms are present (Tobacman 1996; Purcell et al., 1999). The monomers are aligned together in a head-to-tail arrangement, i.e. certain region of C-terminal overlapping with the corresponding region in N-terminal of the adjacent protein. Kobayashi et al. (2008) have shown that carboxypeptidase digestion of this overlapping region invariably weakens the head-to-tail binding and also the association of Tm with actin monomers (Hitchcock-DeGregori, 2000). Further analysis revealed that Ser-283 towards C-terminal most likely makes salt bridges with the N-terminal Lys-7 or Lys-14 of the next Tm molecule. Ser-283 is likely to be phosphorylated by tropomyosin kinase and the phosphorylated Ser-283 substantially increases the numbers of salt bridges and strengthens the head-to-tail interaction. Recently, 2.1 Å resolution crystal structure of Tm C- and N-terminal overhangs was examined (Murakami et al. 2008). As depicted in Figure 1.4, two GCN4 Leu zippers, one each at C- and N-terminal regions, stabilize the dimerization of overlapping regions of adjacent Tm. At N-terminus, Met-1 to Lys-15 splays apart and roles over 1-11 residues of C-terminus, At Tm-N-Tm-C junction three hydrophobic residues of N-terminus, Ile-4, Lys-7 and Met-8 make a hydrophobic hole in which Leu-278 of C-terminus of the next Tm inserts as a knob in “knob-in-hole” packing to create an anchor. Each Tm chain is composed of 284 amino acids, and a tight hydrophobic interaction between the dimmers holds them. The dimmer spans over 7 actin monomers of F-actin microfilament.
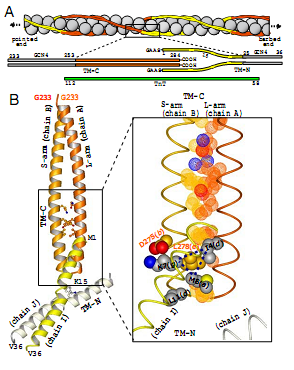
Almost all the tropomyosin chains display an interesting amino acid arrangement (Sodek et al. 1972). There are two series of hydrophobic nonpolar residues each of which repeats at 7 residues apart. Interestingly, the series I residues are at positions 7, 14, 21, 28 … and series II residues at positions 10, 17, 24, 31 …from the C-terminal. In other words, if a sequence of amino acids is a-g, the residues “a” and “d” would be hydrophobic spaced from one another by polar residues “b, c” towards one side and “e, f, g” at the other end. The 7Å crystal structure of Tm shown in Figure 1.4 reveals the periodicities in non-interface positions postulated to be seven quasi-equivalent actin binding sites along the length of one Tm that corresponds to the seven half turns of the supercoil (Whitby and Phillips 2000). The model peptide structures at N and C termini suggest that in solutions the chains are wounded around, but upon crystallization the chains separate from each other in slightly distorted form (Li et al., 2002). Amino acids at positions 167-207 are necessary for myosin S1 “head” binding, whereas at 208-242 make a domain for Tn binding, and thereby they exert the inhibitory role on actomyosin ATPase when cytosolic Ca2+ is depleted at diastole (Hitchcock-Degregori et al. 2002).
The Tm component of thin filament is important for various interactions, like with actin monomers at regular intervals, and with the core domains of Tn (Perry 2001), as well as for the cooperative activation of the muscle thin filament with calcium and myosin. It has been shown in reconstituted skinned muscle fibers that once Tn is present, the affinity of Tm with actin is enhanced by about 80-130 fold (Hill et al 1992). The possible binding sites of Tm with TnT protein of Tn complex is distributed between two regions located at the N and C termini of Tm, which coincides with the head-to-tail overlapping regions. Tm is supposed to interact with TnT at its N-terminus, which is a Ca2+ independent and asymmetric interaction, while the interaction at the C-terminus region of TnT is Ca2+ dependent and symmetric. Besides, Tm also interacts through making ligands with TnT, TnI and TnC (Potter et al 1995; Malnic et al 1998). The schematic representation of Tn-Tm-actin complex is shown in Figure 1.5. As indicated, presence of high Ca2+ perturbs the interaction between Tm and TnT at residues 159-259, whereas strengthens the bonding with TnC (Pearlstone and Smillie 1983). Conversely, when Ca2+ concentration falls, the interaction goes other way around.
The troponin complex
Ebashi pioneered in discovering the troponin as the calcium regulated protein complex with unique regulatory property in muscle contraction (Ebashi, Ebashi et al., 1967). The Tn is a hetero-trimeric-protein complex constituted by three interacting subunits, namely TnC (~18.3 kDa), TnI (~23.5 kDa) and TnT (~38 kDa). After repeated trials, a high resolution crystal structure for human cardiac Tn was deciphered and modeled under Ca2+ saturated conditions, which corresponds to the contractile state (systole) of the myofilaments (Takeda et al 2003). The X-ray crystal structure of Tn core is shown in Figure 1.6. The core domain can be divided into two sub-domains, the regulatory head and the IT region. The N-terminus of TnC and a “switch region’, located towards the C-terminal of TnI, are arranged in anti-parallel orientation, and they constitute the regulatory head. The oppositely tuned interaction of TnI and TnC towards regulatory head involves the two EF hands (helix-loop-helix) of B and C helixes of Ca2+-bound TnC in “open” conformation and the “switch region” of TnI (residues 116-131; helix H3(1). Ahead of this, the 150-159 residues of TnI also interact with the Ca2+-bound “open” hydrophobic patch of the N terminus of TnC. Besides, the inhibitory segment of TnI (residues 104-115) exists in an ordered loop that also interacts with TnC. TnI at the C-terminus protrudes as another α-helix (residues 166-188; helix H4(1), and apparently in the crystalline state does not directly associate with the rest of the Tn complex, though, contribute immensely towards inter-domain interactions and Ca2+ sensitivity. In the IT region three parallel helical domains of TnI and TnT wind each other and hold the C-terminus domain of TnC. These helixes are the N-terminal helix of TnI (residues 8-48; H2(1), the TnI residues 90-135 (H1(1), and TnT residues 226-271 (H2(T2). TnT residues 204-220 upstream from the coiled coil form the α-helix, represented as H1(T2). While one end of the H2(T2) helix make coiled-coil which interacts with TnI (H1(1), the other end of H2(T2) associates with the C-terminus EF hands (helix-loop-helix) of E and F helixes of Ca2+-bound TnC (Takeda et al. 2003). TnT1 segment is normally excluded from being able to crystallize because it remains in aggregate rather than in lattice form (King et al 2005). The Tn complex displays unique signature amino acid sequences that constitute hydrophobic, acidic, basic and Ca2+ binding domains, dynamism in inter-and intra-domain interactions, and post-translational modifications like phosphorylation.
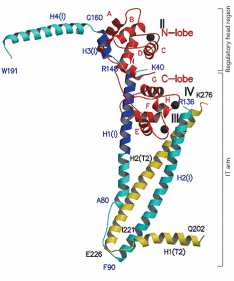
TnC – Apart from calmodulins, in sarcoplasma TnC is the other prominent Ca2+-binding protein. Two relatively similar isoforms of TnC are expressed in vertebrate genome: the slow skeletal/cardiac muscle TnC (cTnC) and fast skeletal muscle TnC (sTnC), which exhibit low and high Ca2+ affinities, respectively, due to alterations in calcium binding pockets. The structure of sTnC, as determined by X-ray crystallography (Herzberg and James 1988; Houdousse et al 1997) and NMR spectroscopy (Slupsky and Sykes 1995), reveals it to be 76 Å long and dumb bell shaped protein of ca. 18 kDa mass. The protein is considered to be highly acidic owing to the presence of glutamate and aspartate residues higher than average proteins (Zot and Potter, 1987). Although cTnC and sTnC possess 161 and 160 amino acid residues about 54 residues are different, and this accounts for only 65% similarity between them. There exist two globular domains in structure of both TnC’s corresponding to the N- and C-termini of the proteins, separated by a long helical linker.
TnC belongs to the tetraEF class of helix-loop-helix proteins that are known to bind to Ca2+. Towards the N-terminus loops are found between helixes A and B (site I) and C and D (site II), and at C-terminus between E and F (site III) and G and H (site IV). Sites III and IV nonspecifically occupy a divalent cation predominantly Mg2+ or Ca2+, whose binding affinity is high, and in physiological conditions it stays in bound state. The cations in this structural region maintain the integrity of Tn and prevent TnI and TnC dissociation (Sin, Fernandes et al., 1978). In contrast, sites I and II in case of sTnC and only site II in cTnC bind to Ca2+ at much weaker affinities (Gorden et al. 2000). For the inactivation of site I in cTnC single insertion and two substitution of amino acids is sufficient – Val insertion at residue 28, and Asp replacement with Leu at residue 29 and Asp with Ala at residue 31. This brings down Ca2+ affinity by eight-fold compared to sTnC and correspondingly weakens the contractile force and overall efficiency. Ca2+ binding employs 12 residues in the EF helix-loop-helix structures. Within this signature binding site 1, 3, 5, 7, 9 and 12 amino acid residues make coordination bond with Ca2+ using the side chain carboxylate oxygen. In apostate the two N-terminal domains in sTnC (sites I and II) and only one in cTnC (site II), which contain hydrophobic amino acid residues, remain closed, i.e. buried in the structure, whereas upon Ca2+ binding the corresponding domains dramatically open up, exposing the hidden hydrophobic residues. The hydrophobic region attracts the TnI, pulling the protein at C terminus away from actin, thus enabling the myosin S1 “heads” to bind to the actin for onward contraction process. Besides, such conformational change of TnI hydrophobic residues also enables transmission of Ca2+ mediated signals, and this ensures overall stability of Tn complex.
TnI – This subunit of Tn complex is also known as the inhibitory subunit, and in vitro it is shown to inhibit actomyosin ATPase activity, which is the driving force for sliding of muscle microfilaments. There exist three structural isoforms of TnI in vertebrates: the cardiac TnI (cTnI), the fast twitch skeletal TnI (fsTnI) and the slow twitch skeletal TnI (ssTnI). The fundamental difference between the human cTnI and the skeletal isoforms of TnI’s is an additional 27 to 33 amino acid residues towards the N-terminus tail, which composes the total 209 residues for cTnI, while for ssTnI it is 186 and for fsTnI only 181 (Li et al. 2004). This extended region in cTnI includes a region rich in acidic residues and another extensive phosphorylation region that interacts with C-domain of cTnC (Perry 1999). The common functions and structures between skeletal and cardiac TnI isoforms can be seen towards the C-terminus of the protein due to structural dissimilarity, but not at N-terminus. It has been unequivocally proven that TnC and TnI at 1:1 ratio are capable of alleviating the inhibitory action of Mg2+ actomyosin ATPase by solely TnI. TnI is also assumed to stabilize the entire troponin complex together by Ca2+ regulated binding with TnC and TnT, and it facilitates the Ca2+-dependent binding of TnT with actin. In addition to the above functions, the residues 128-147 in cTnI, and 96-115 in fsTnI or ssTnI are responsible to prevent binding of myosin S1 “head” with actin in presence of Ca2+.
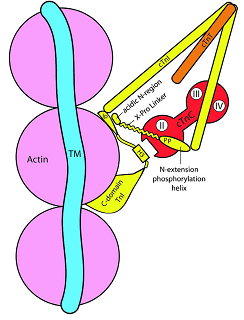
Structurally and functionally the N-terminal extended region of cTnI has gathered attention recently due to its role in Ca2+-dependent contraction in co-ordination with cTnC, which has implications in β-adrenergic signaling pathways in cardiac pathophysiology. Sadayappan et al. (2008) segregated the N-terminus of cTnI into three regions: an acidic N region with single turn of helix (1-10 residues), followed by a polyproline rich domain (11-20 residues) and another helix with property of extensive phosphorylation (21-30). With β-adrenergic signaling the Ser23/24 is phosphorylated with AMP dependent protein kinase A (PKA). This phosphorylation results in Ca2+ desensitization, i.e. reduced affinity of Ca2+ to the sole binding pocket in cTnC. This is executed by a conformational change in the phosphorylated cTnI. The NMR data reveals that residues 19-30 are minimum required for a weak interaction of N-terminus of cTnI and N-terminus of cTnC in proximity to the inactive Ca2+-binding site I. This interaction greatly affects the Ca2+ affinity at site II in N-terminus region of TnC. In the non-phosphorylated state, the N-extension of cTnI contacts the N-terminus site I of cTnC and influence the site II Ca2+ binding. Upon PKA-mediated Ser23/24 phosphorylation, the above affinity weakens and N-terminal of cTnI bends such that the acidic region makes electrostatic interaction with the inhibitory region of cTnI. Consequently, Ca2+ affinity of cTnC also decreases. In a cTnI protein construct in which 2-11 residues were deleted, the remaining cTnI was neither capable to interact with N-terminal of cTnC, nor was able to block the inhibitory site of cTnI. An overall manifestation of phosphorylation on cTnI-cTnC interaction was detailed by Solaro et al. (2008), which re-affirmed the prevalence of PKA-mediated Ser23/24 phosphorylation as overall modulator of cTnC-Ca2+ binding affinity, follow-up signal transduction to cTnT and overall cardiac contractility. Accordingly, with phosphorylation, the affinity of N-terminus of cTnI is further weakened with N-terminal site I of cTnC causing Ca2+ desensitization or greater Ca2+ release from cTnC. Eventually, the acidic N-terminus of cTnI is positioned such that it binds to the inhibitory region of cTnI. As a result the interaction of the inhibitory region and actin is weakened with alteration in cross-bridge reaction. Phosphorylation of Ser-42/44 towards N-domain and Thr-143 in the inhibitory region in cTnI is achieved by protein kinase C (PKC). A larger implication of this modification can be seen on decreased maximal actomyosin Mg2+-ATPase. Overall, the PKA and PKC mediated phosphorylation at the respective positions in cTnI have quite an opposite response on cross-bridge formation. Figure 1.7 describes the structural domains of extended cTnI N terminus.
TnT – It is an elongated highly symmetrical rod shaped protein of approximately 18 nm responsible for binding with TnI, TnC and Tm. TnT can be easily cleaved with chymotrypsin into C- and N-terminal fragments. Essentially, the N-terminus of TnT binds to the C-terminus of Tm, especially at the overlapping region of head-to-tail association of C- and N-terminus overlapping region of Tm. The C-terminus of TnT interacts with TnI, TnC and Tm. In vertebrates, TnT is expressed in three isoforms – cardiac (cTnT), fast twitch skeletal (fsTnT) and slow twitch skeletal (ssTnT). The length of TnT is variable in range of 160-300 amino acid residues, hence expectedly most of the region in vertebrate TnT is not conserved, though the main cTnT isoform in normal adult heart is composed of 288 amino acids, and upon proteolytic cleavage it can been separated in two fragments – an extended amino-terminal T1 domain (cTnT1, residues 1–181) and a globular carboxy terminal T2 domain (cTnT2, residues 182–288). The variation within the cTnT even in one individual is expected even though there is only one gene, TNNT2, expressing all the variants. This happens because alternate splicing sites present in pre-mRNA get processed into several cTnT mRNA species (Mesnard et al 1995). Interestingly, cTnT is recognized as highly charged protein; the N-terminus region comprises the 30% acidic residues whereas the C-terminus has predominantly the 20% basic residues (Pearlstone, Carpenter et al., 1976). At physiological pH and low ionic strength this highly polar protein hardly stays in soluble form. The fragment responsible for Tm binding towards the N-terminus, also called the TnT1, is vested by a patch of amino acid residues 71-151, and this binding is considered Ca2+-insensitive and deleting 45 residues do not change cTnT’s response towards Ca2+. On the other hand, the Ca2+-sensitive binding of TnT with Tm is attributed to the fragment in the C-terminal fragment, also known as TnT2.
In a recent 2.9 Å resolution crystal analysis of skeletal Tn:Tm complex (Murakami et al. 2008), residues 66 to 99 in TnT was found to be crucial for binding with overlapping region of Tm. The residues 58 to 112 in TnT is highly conserved. The binding partially unfolds the Tm, but rotation angles vary in N- and C-termini in presence of Ca2+. Considering Tm surrounding I-VII actin monomers in F-actin, the N-terminus region covering V-VII actin moves deeper into the actin grooves yielding a “closed” state, to which myosin heads bind weakly. Conversely, C-terminus of Tm covering I-IV actin residues fail to slide much resulting in generation of “open” state of actin, to which myosin heads bind strongly. Surprisingly TnT has practically no influence on the inhibitory action of TnI on actomyosin ATPase, but as shown above, do influence the numbers of open and closed states of cross bridges between actin and myosin, and this regulates overall contractibility in presence of Ca2+.
Calcium mediated regulation of myocardial activities
According to Bers (2008), in myocytes calcium exerts diverse signaling events that include:
- mediation of normal and arrhythmic action potential shape by regulating calcium channels and cation exchangers,
- connection between the electrical excitation signals and contraction through intracellular calcium transients,
- contraction of actomyosin myofilaments,
- energy turnover,
- apopototic cell death,
- gene expression through calmodulin-dependent activation of transcriptional factors.
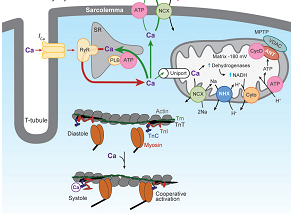
Some calcium regulated or dependent processes are interrelated and malfunction of one or the other signaling events lead to co-ordinated structural, mechanical and electrical cardiac disfunctions. Most of the signaling is mediated through the specific calcium-binding proteins, namely calmodulins and cTnC. Excitation-contraction coupling is a process in which electrical impulses are translated to cross-bridge activities in the muscle fibers, actin and myosin. In mammalian heart, depolarization increases the cytoplasmic calcium concentration (10-20 µmol Ca2+/g fresh wt) by activating the L-type Ca2+ channels, also known as dihydropyridine receptors (DHPR) present on sarcolemma membrane. For an effective contractile action to take place a minimum 30-50 µmol Ca2+/g fresh wt is required in cytoplasm. The calcium homeostasis is maintained by simultaneous expulsion of calcium through Ca2+-mediated activation of sarcoplasmic reticulum Ca2+ release channel or ryanodine receptors (RyR) by a process known as “calcium-induced calcium release”. Sarcoplasmic reticulum is the calcium storehouse and upon RyR activation the calcium starts to release in the cytosol. Collectively, the calcium influx and cytosolic release channels comprise a local Ca2+ signaling complex called coupon. The cytoplasmic calcium builds up to 200-400 µmol Ca2+/g fresh wt, a level sufficiently high to bind to the cTnC, and to activate the contractile activity of the myofilaments through stimulating myosin ATPase. The process of relaxation is accompanied by reduction in cytoplasmic Ca2+ level and dissociation of cTnC-bound Ca2+. This is accomplished by two routes: re-absorption of Ca2+ in sarcoplasmic reticulum by the Ca2+ATP-dependent pump facilitated by calcium-calmodulin-dependent protein kinase (CaMKII) which phosphorylates the phospholamban (PLB). The kinase also induces the release of Ca2+ through RyR system for diastole that activates the expulsion of Ca2+ from sarcomere through Na+/Ca2+ exchangers. A minor fraction of Ca2+ is removed from cytosol using the sarcolemmal Ca2+ ATPase and mitochondrial transporters. The sarcoplasmic Ca2+ homeostasis is presented in Figure 1.8.
The calcium regulation of Tn complex dynamics and eventual Tm.actin interaction has been reviewed by Stehle et al. (2007). Accordingly, an increase in cytosolic Ca2+ makes the site II of cTnC filled with Ca2+ and this, in turn, activates the contraction process. The calcium binding sites take an “open” configuration, thereby attracting cTnI, in particular the “switch region”. Consequently, cTnI C-terminus is pulled away from its actin binding sites, and Tn-Tm complex rolls deeper into the grooved positions of actin polymer (Lehman, Hatch et al., 2000). This enables myosin heads to sit on the positions in actin and cross-bridge is formed (Farah and Reinach, 1995; Solaro and Rarick, 1998; Squire and Morris, 1998; Gordon, Homsher et al., 2000). A cross-bridge further deepens the positioning of Tn-Tm in actin grooves and a co-operative manner helps cross-bridge formation in adjoining actin sites, and Ca2+ binding to more apo-cTnC proteins. Figure 1.9A is a cartoon representation of the conformational changes that occur during muscle contraction (Li, Wang et al. 2004) and in Figure 1.9B the individual protein components’ movements in cardiac sarcomere.
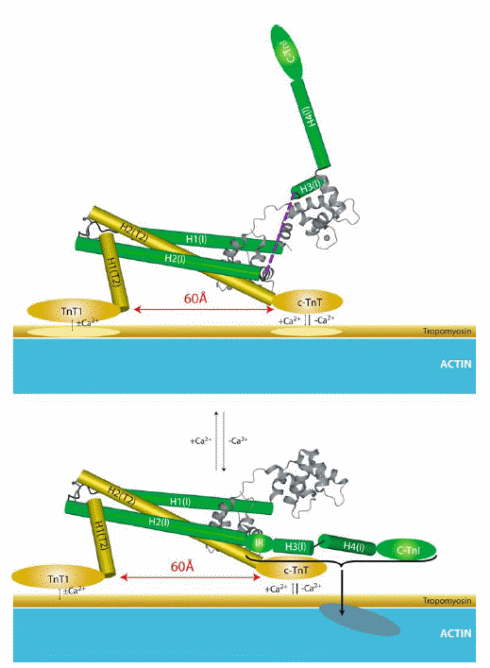
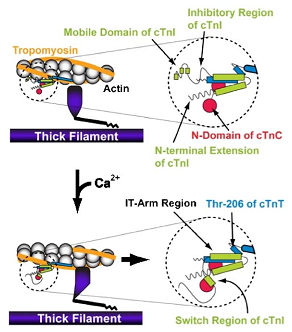
It is normally experienced that during relaxation there is a time lag even though the cytosolic Ca2+ concentration sharply falls. Two fundamental events facilitate the relaxation upon calcium depletion. First, PKA-mediated cTnI phosphorylation brings down the affinity of cTnC towards Ca2+, and this automatically transforms the site II back in “closed” position, thereby releasing the previously bound “switch region” of cTnI, and the entire C-terminus of TnI. The actin binding sites are then recaptured and this brings the Tn-Tm complex towards surface of actin molecules from the grooves. Consequently, myosin heads are detached from actin to break contractility. Secondly, frequency dependent acceleration of relaxation is facilitated by CaMKII-dependent PLB phosphorylation at Thr-17. This enables rapid intake of Ca2+ in sarcoplasmic reticulum, thereby speedily depleting the cytosolic Ca2+ concentration even further.
Regulation Muscle Contraction
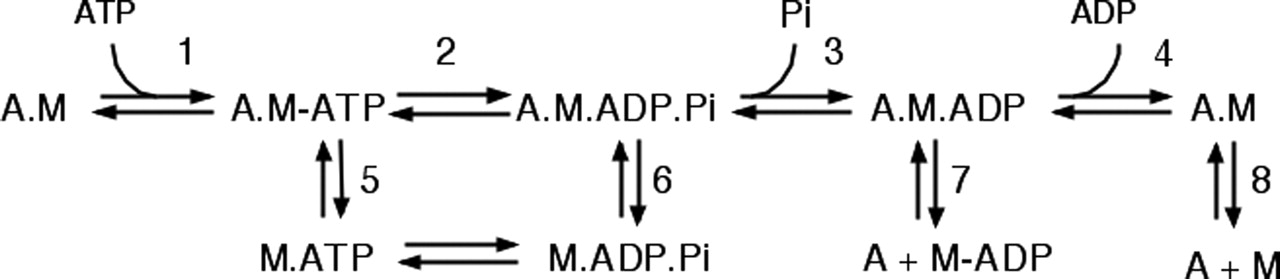
Since past half century several theories, as summarized in Figure 1.11, were put forth and extensive investigations were carried out to explain contraction mechanistic (Geeves and Holmes, 1999; Holmes and Geeves, 2000). Before 1970 the popular sliding filament theory explained how a cycling interaction of actin and myosin can move, but later the model was updated and a role of hydrolysis of ATP by myosin and myosin-linked alternating conformational changes in complementary actin sites were related to the sliding of filaments. This “cross bridge model” was later improvised by Spudich and co-workers (Spudich et al 1994), who suggested that the conformational change in the cross bridge also involves a swing of the myosin heads by “lever arm mechanism”.
The entire mechanistic was reproduced in test tube with reconstituted skinned muscle filaments, and scheme 1.1 depicts how Tm-actin-activated myosin Mg2+ ATPase maneuvers the cross bridge cycling. The first step, as shown by Criddle and co-workers using fluorescent titration of pyrene skeletal actin and myosin-S1 “head”, has been a tight binding of myosin and actin in the order of magnitude of 9.1X108 M-1 (Step 8) ( Criddle et al. 1985). The later steps were, instant ATP binding with S1 (Step 1) that causes rapid dissociation of myosin from actin (Step 5) followed by fast ATP hydrolysis to ADP and Pi at maximum rate of around 117 s-1 in case of skeletal muscle. ATP hydrolysis can also take place while actin is still bound to myosin (Step 2) (Sleep and Hutton 1978). The myosin.ADP.Pi then rebinds to actin (step 6) and this dynamic complex undergoes a slow transition to a stable high affinity actin-myosin complex by releasing Pi (the rate limiting step 3) at a rate of 75 and 20 s-1 respectively for skeletal and cardiac muscles. The bound ADP then dissociates, leaving actinmyosin complex (Step 4) at a much slower pace, the maximum being ~400 s-1 for skeletal and 100 s-1 for cardiac muscles. Upon release of cTnI “switch region” and attachment of myosin heads to the actin in presence of Ca2+ ATP hydrolysis into ADP and Pi as shown in scheme 1.1 takes place. This triggers the myosin heads to anchor, forcing the actin and myosin to slide over each other to generate the power stroke. For a new contractile cycle to start, the myosin head must bind to new ATP molecule. Conversely, with fall in cytosolic Ca2+, ATPase is inhibited from attachment of cTnI “inhibitory region”, and the muscle relaxes. The responsiveness of thin filament to both Ca2+ and myosin heads is determined by Tm. It is becoming clear that rather than only the cTnC or cTnI protein it is the conformational change of the entire thin filament, as proposed by the classic steric blocking hypothesis (Lehrer, 1994; Lehrer and Geeves, 1998), that leads to muscle relaxation.
Davis & Tikunova (2008) outlined modulation of calcium affinity with cTnC as the complexity of the cardiac muscle increases due to reconstitution of components of thin filaments. The isolated cTnC exhibits affinity of >1000 s-1 which drastically lowers down to ~120 s-1 upon reconstitution with TnI and even further to ~35 s-1 with TnT. Addition of actin and Tm though slightly increased this rate to ~105 s-1, further complementation with myosin S1 heads reduce it further to ~13 s-1. Hence, consideration that cTnC alone is the Ca2+ regulatory unit for muscle contraction may be oversimplified, as the dissociation rate is too fast to channel the effect at myosin level. Thus, it seems that the Ca2+ dissociation is a co-ordinated action of the entire complex, TnC.TnI.TnT.actin.myosin S1 “head”. A mutation in any one of the components is expected to affect Ca2+ sensitivity and muscle contractility.
The thin filament regulation is a cooperative allosteric three state mechanism accounting for both Ca2+ and myosin S1 “head” activation (Lehrer and Geeves, 1998). Moreover, McKillop and Geeves (McKillop and Geeves, 1993) proposed a Ca2+ dependent equilibrium between the three states (Scheme 1.2):
- The blocked state – dominates in absence of Ca2+ when myosin cross-bridges do not interact with actin; low ATPase activity.
- The closed state – dominates in presence of Ca2+. Here the cross-bridges weakly bind to actin; ATPase activity is still low.
- The open state – dominates in the presence of myosin heads, cross-bridges bind to actin to enable strong actinmyosin binding; myosin ATP breakdown activated producing high force.
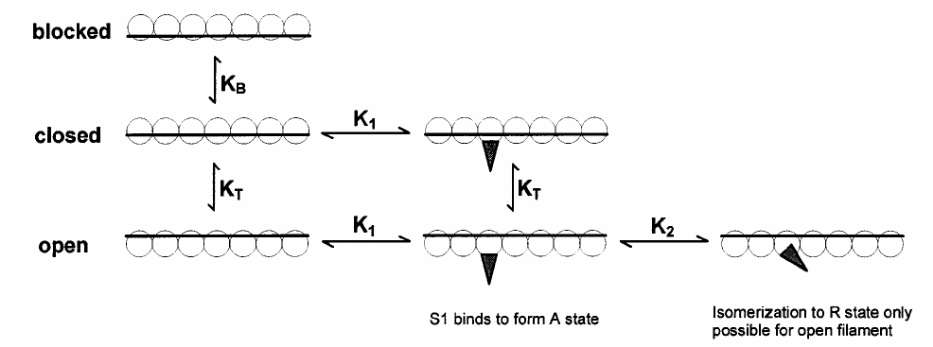
The three binding state transitions are cooperative, determined by the size of cooperative unit ‘n’ (Geeves and Lehrer, 1994), equilibrium between blocked and closed states “KB”, and equilibrium between closed and open states “KT” (Maytum et al 1999). In the open state, cross-bridge cycling and contraction is achievable. Cooperativity can be negative at low Ca2+ state (relaxation/diastole) where Tn serves as an allosteric inhibitor, or positive at high Ca2+ state (contraction/systole) where Ca2+ and strong myosin binding residues are the allosteric activators.
Pathophysiology of cardiomyopathies due to mutations in sarcomeric proteins
Cardiovascular diseases are the leading cause of worldwide illness and mortality, including in US and U.K. In US alone this accounts for 1 million deaths annually (40% of all causes together). Cardiomyopathy, a complex pathophysiological condition of myocardial dysfunction can be categorized into 4 main types (Morimoto 2008; Ashrafian & Watkins 2007): hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), restrictive cardiomyopathy (RCM), and arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C). For ARVD/C no sarcoplam protein aberration has been assigned. HCM and DCM increase the myocardial thickness and affect the ventricular volume. HCM is manifest as thickening (hypertrophy) of ventricular wall and the interventricular septum that decreases the ventricular space. In HCM, systolic pressure slightly enhances, while diastolic functions are perturbed due to interstitial fibrosis, disarray in myocytes and hypertrophy itself, resulting in heart failure and early age sudden cardiac deaths. DCM also increases ventricular volumes and thickening of wall but is manifest with systolic dysfunction, leading to heart failure and sudden deaths. RCM also displays more severe HCM-like symptoms, and is characterized by restricted diastolic function, reduced left ventricular filling and diastolic volume. The disorders could be acquired or inherited through specific autosomal (dominant or recessive), or X-linked inherited traits. HCM is a complex genetic disorder involving 9 sarcomeric genes, including TNNT2 for cTnT, MYBPC3 for myosin binding protein C, MYH7 for β-myosin heavy chain, TPM1 for α-tropomyosin and TNN13 for Tn1, and >450 mutations, mainly missense. The defective sarcomere proteins eventually disrupt the developmental functions like cellular differentiation, growth and cytoskeletal organization. It was hypothesized that sarcomere mutations deficit energy usage, leading to cyto-architectural disorganization. Aberrant signaling of ATP:ADP imbalances induce AMP-activated protein kinase and decrease Ca2+ reuptake from sarcoplasmic membrane, which manifests in hypertrophy. While HCM is principally linked to genetic inheritance of mutated genes, DCM is associated with many acquired characters like myocardial ischaemia (insufficient blood flow), inflammation, infection, pregnancy, alcohol, autoimmune disorder and intoxication, with 20-50% cases linked to inheritable traits. DCM may appear as secondary manifestation in many skeletomuscular disorders other than sarcomere proteins, like Duchenne’s and Becker’s muscular dystrophies, caused by mutations in the Dystrophin gene with no direct relationship to cardiac function. Typically, about 6 sarcomere genes common for HCM are also mutated for DCM. In fact mutations in common sarcomere genes may have exactly opposite disease manifestations of DCM and HCM. Histological appearance of HCM heart (myofiber disarray) is absent in DCM; in vitro the HCM and DCM mutants behave antagonistic, e.g. DCM mutation depresses myofibrillar function whereas HCM mutations enhance, and unlike HCM mutations, in DCM reduce Ca2+ sensitivity and thin filament activation take place and apoptosis rates leading to stressed myocyte death are higher. Notwithstanding about 5% of HCM patients display DCM like characteristics.
cTnT mutations and HCM/DCM phenotypes
Morimoto (2008) compiled troponin and other sarcomere protein-linked cardiomyopathic conditions. Twenty seven cTnT linked mutations in TNNT2 gene were associated with HCM as listed in Table 1.1. Reconstruction of the crystal structure of the core domain (C.I.T2 complex) from human cardiac Tn complex is shown in Figure 1.12. Most mutations were found in non-crystallizing mobile functional domain of cTnT, such as the Tm-actin, cTnI or cTnC binding sites, located outside the core domain. Surprisingly, cTnT patients do not present severe cardiac hypertrophy but still incidences of sudden mortality remain high. Life expectancy is ~35 years for patience with missense mutations, S179N, R92Q, deletion mutation ΔE160, and splicing mutation 16G1→A. Using rabbit or rat skinned myofibrils composed with human cTnT mutations S179N, and R92Q/W/L, different investigators reported elevated myofilament Ca2+ sensitivity, without making any difference to maximum force generation and myosin ATPase activity. A transgenic mice expressing human R29Q cTnT mutation exhibited enhanced contractility, and diastolic abnormalities, with myocytes displaying shortened sarcomere lengths, increased sarcometric activation and reduced relaxation, suggestive of increased Ca2+ sensitivity.
Table 1.1: TnT mutations relative to the functional domains. Mutations investigated in this thesis are in bold.
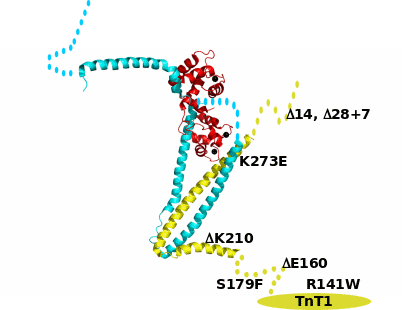
The first cTnT mutant, ΔK210 in TNNT2 gene, was identified to be responsible for two distinct familial DCM. Decrease in Ca2+ sensitivity for force generation and ATPase activity in cardiac myofibrils were the primary pathogenic response in skinned cardiac fibers. Other mutations, R141W, R131W, R205L and D270N, also caused subdued Ca2+ sensitivity without affecting maximum force generation, besides enhancing the cTnC.α-Tm affinity. The patients died early with heart failure. Presently six cTnT mutations are attributed to DCM. A knock-in mice with K210 codon deletion analogous to human DCM conditions displayed swollen heart, reduced Ca2+ sensitivity, and premature deaths. Administration of a Ca2+ sensitizer, pimobendan, completely reversed all the DCM characteristics. Lombardi et al. (2008) constructed two mouse models with cTnT mutations, R92Q and R141W, which are prevalent in HCM and DCM patients, respectively. Indeed the mice phenotypes resembled to the human cardiomyopathy. Upon autopsy R141W mouse showed dilated heart with lower diastole. Conversely, constricted heart and high systole were characteristics of R92Q mouse. Ca2+ sensitivity of myofibril and myosin ATPase responded opposite in the two transgenic animals. The binding properties of cTnT with other thin filament proteins were examined in two mutated mice and it was revealed that R141W displayed higher affinity for cTnC, α-Tm and α-actin, whereas cTnI binding was more with cTnT R92Q. Such contrasting affinities among the thin filament proteins could be one of the reasons for HCM and DCM phenotypes.
The approach of mimicking the human genome mutations using reconstituted animal skinned myofibril led to some erroneous conclusions. This issue was raised by Chang et al. (2008) in cTnT S179N mutation, known for HCM phenotype. This mutation had induced ventricular tachycardia (abnormal pulse) even in absence of hypertrophy. Thus, more than anatomical changes in heart the arrhythmias were the cause for diastole dysfunction and sudden cardiac deaths. Fiset & Giles (2008) attempted to relate Ca2+ sensitivity and ventricular electrophysiology in relation to such cTnT mutations, and concluded that Ca2+ desensitization is positively correlated to ventricular tachycardia.
Changes in intracellular Ca2+ concentration would affect outward transport of Ca2+ using Na+/Ca2+ exchangers and this electrogenic transporter, in turn, can modulate ventricular excitability. A comprehensive assessment of Ca2+ sensitization and arrhythmias in HCM-linked cTnT transgenic mice was carried out by Baudenbacher et al. (2008), who used three human missense mutants, S179N, F110I and R278C, which also exhibited decreasing trend in Ca2+ sensitization in human cardiac myofibers. There was no evidence of fibrosis or hypertrophia in transgenic mice, but in skinned fiber preparations the Ca2+ sensitivity was in order of S179N>F110I>R278C. It was concluded that, independent of any anatomical changes in heart, cTnT mutations in the same graded order caused risk of ventricular tachycardia by changing the ventricular action potential shape, shortening retractile periods between action potentials, beat to beat T wave shape and amplitude alterations, and faster than normal dispersal of ventricular conduction. In a similar investigation on cTnT ΔK210 mice, which should show DCM phenotype, the death was quite rapid due to arrhythmias and induction of long QT (QT gap increases due to delayed repolarization), associated with increased phosphorylation of RyR and PLB (Solaro 2007).
Another cTnT mutant in animal model, Δ160E with HCM characteristics, was also found to alter the Ca2+ fluxes and induced ventricular arrhythmic action potential. The nature of replacing amino acids in single point mutation also profoundly affects the type and severity of the disease. E.g. cTnTR92 acquiring W develop negligible or mild hypertrophy but such patients experience sudden cardiac deaths and the disease progressed from HCM to DCM; upon replacement with L the carriers developed acute hypertrophy but no sudden deaths, and if it were Q then the response goes more like S179N patients and ventricular beats and rhythms got perturbed leading to sudden cardiac deaths (Chang et al. 2008).
Taken together, the cTnT – S179N, R92Q/W/L, F110I, R278C, R141W, R131W, R205L, D270N, ΔK210, Δ160E, 16G1→A mutations have produced inconsistent results and sometimes the in vivo implications of such mutations could not be explained from results of reconstituted skinned microfibers. Potential environment surrounding ventricular sarcomere is different from the isolated muscle fibers, the constitution of cTnT isoform under given diseased condition might be different, the interaction with other thin filament proteins may vary and the electrogenic stimulation may exert its impact. There is still no clear understanding as to why cTnT mutations alter Ca2+ sensitivity to maximum force generation and ATPase activity. Likely, binding of cTnT with actin, Tm and cTnC/cTnI may be affected. Possibly cTnI phosphorylation gets modified and this affects Ca2+ sensitivity of cTnC and/or movement of the inhibitory arm away or close to actin and other cTnC configurational changes. Tm affinity with actin can be jeopardized such that actinmyosin can not transit from “closed” to “open” state and thus ATPase activity and the maximum force generation may get perturbed.
Programme of Research
Looking into the prevailing ambiguity in relating the cTnT mutations and type and magnitude of cardiomyopathy, it was felt necessary to investigate precisely how mutations affect overall Ca2+ sensitivity, maximum force generation and ATPase activity in reconstituted cardiac muscle fibers, based on already available procedures already standardized for skeletal muscle. The objective of this project is to examine the effect of seven selected cTnT mutations causing human DCM and HCM on detailed kinetics of:
- Thin filament switching,
- Calcium binding association and dissociation.
Reference List
Ashrafian H & Watkins H 2007, ‘Reviews of Translational Medicine and Genomics in Cardiovascular Disease: New Disease Taxonomy and Therapeutic Implications Cardiomyopathies: Therapeutics Based on Molecular Phenotype’, Journal of the American College of Cardiology, vol. 49, no. 12, pp. 1251-1264.
Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD & Knollmann BC 2008, ‘Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice’, Journal of Clinical Investigation, vol. 118, pp. 3893-3903.
Bers DM 2008, ’Calcium Cycling and Signaling in Cardiac Myocytes’, Annual Reviews of Physiology, vol. 70, pp.23–49.
Chang AN, Parvatiyar MS & Potter JD 2008, ’Troponin and cardiomyopathy’, Biochemical and Biophysical Research Communications, vol. 369, pp. 74–81.
Davis JP & Tikunova SB 2008, ‘Ca2+ exchange with troponin C and cardiac muscle Dynamics’, Cardiovascular Research, vol. 77, pp. 619–626.
Fiset C & Giles WR 2008, ‘Cardiac troponin T mutations promote life-threatening arrhythmias’, Journal of Clinical Investigation, vol. 118, pp. 3845–3847.
Gorden AM, Homsher E & Regnier M 2000, ’Regulation of Contraction in Striated Muscle’, Physiological Reviews, vol. 80, pp. 853-924.
Kobayashi T, Jin L & Tombe PP 2008, ‘Cardiac thin filament regulation’, Pflugers Archives – European Journal of Physiology, vol. 457, pp. 37–46.
Li MX, Wang X & Sykes BD 2004,’Structure based insights into the role of troponin in cardiac muscle pathophysiology’, Journal of Muscle Research and Cell Motility, vol. 25, pp 559-579.
Lombardi R, Bell A, Senthil V, Sidhu J, Noseda M, Roberts R, & Marian AJ 2008, ‘Differential interactions of thin filament proteins in two cardiac troponin T mouse models of hypertrophic and dilated cardiomyopathies’, Cardiovascular Research, vol. 79, pp. 109-117.
Morimoto S 2008, ‘Sarcomeric proteins and inherited cardiomyopathies’, Cardiovascular Research, vol. 77, pp. 659–666.
Murakami K, Stewart M, Nozawa K, Tomii K, Kudou N, Igarashi N, Shirakihara Y, Wakatsuki S, Yasunaga T & Wakabayashi T 2008, ‘Structural basis for tropomyosin overlap in thin (actin) filaments and the generation of a molecular swivel by troponin-T’, Proceedings in National Academy of Science USA, vol. 105, no. 20, pp. 7200-7205.
Sadayappan S, Finley N, Howarth JW, Osinska H, Klevitsky R, Lorenz JN, Rosevear PR & Robbins J 2008, ‘Role of the acidic N’ region of cardiac troponin I in regulating myocardial function’, FASEB Journal, vol. 22, pp. 1246–1257.
Sodek J, Hodges RS, Smillie LB & Jurasek L 1972, ‘Amino-Acid Sequence of Rabbit Skeletal Tropomyosin and Its Coiled-Coil Structure’, Proceedings in National Academy of Science USA, vol. 69, no. 12, pp. 3800-3804.
Solaro RJ 2007, ‘Translational Medicine With a Capital T, Troponin T, That Is’, Circulation Research, vol. 101, pp. 114-115.
Solaro RJ, Rosevear P & Kobayashi T 2008, ‘The unique functions of cardiac troponin I in the control of cardiac muscle contraction and relaxation’, Biochemical and Biophysical Research Communications, vol. 369, no. 1, vol. 82–87.
Stehle R, Iorga B & Pfitzer G 2007, ‘Calcium regulation of troponin and its role in the dynamics of contraction and relaxation’, American Journal of Physiology- Regulatory Integrative and Comparative Physiology, vol. 292, pp. R1125–R1128.